Пороки развития кожи
Последнее обновление: 21.02.2021
Пороки развития стойкие морфологические изменения органа, системы или организма, которые выходят за пределы вариаций их строения и возникают внутриутробно в результате нарушений развития зародыша или (много реже) после рождения ребенка как следствие нарушения дальнейшего формирования органов. К порокам развития относятся аплазия, врожденные гипоплазия и гиперплазия, , гетеротопия, гетероплазия, эктопия, В зависимости от этиологии все П. р. разделяют на возникшие в результате генных мутаций (мономутантные пороки развития); хромосомных и геномных мутаций (хромосомные синдромы); комбинированного воздействия генных мутаций и факторов внешней по отношению к зародышу среды (мультифакториальные пороки развития); тератогенных факторов (группа бластопатий, эмбриопатий и фетопатий).
Не существует единой классификации и сколь-либо удачного определения пороков кожи, и это не удивительно, т. к. большинство кожных заболеваний так или иначе являются генетически детерминированными и могут быть отнесены к порокам развития. В Международной Классификации Болезней 10 пересмотра так же нет какой-либо чёткой последовательности в подходе к систематизации этих болезней.
Международная классификация болезней 10-го пересмотра (МКБ-10)
Класс 17 Врожденные аномалии [пороки крови], деформации и хромосомные нарушения
Q80-Q89 Другие врожденные аномалии [пороки развития]
Q80 Врожденный ихтиоз
- Q80.0 Ихтиоз простой
- Q80.1 Ихтиоз, связанный с Х-хромосомой [Х-сцепленный ихтиоз]
- Q80.2 Пластинчатый [ламинарный] ихтиоз
- Q80.3 Врожденная буллезная ихтиозиформная эритродермия
- Q80.4 Ихтиоз плода ["плод Арлекин"]
- Q80.8 Другой врожденный ихтиоз
- Q80.9 Врожденный ихтиоз неуточненный
Q81 Буллезный эпидермолиз
- Q81.0 Эпидермолиз буллезный простой
- Q81.1 Эпидермолиз буллезный летальный
- Q81.2 Эпидермолиз буллезный дистрофический
- Q81.8 Другой буллезный эпидермолиз
- Q81.9 Буллезный эпидермолиз неуточненный
Q82 Другие врожденные аномалии [пороки развития] кожи
- Q82.0 Наследственная лимфедема
- Q82.1 Ксеродерма пигментная
- Q82.2 Мастоцитоз
- Q82.3 Недержание пигмента [incontinentia pigmenti]
- Q82.4 Эктодермальная дисплазия ангидротическая
- Q82.5 Врожденный неопухолевый невус (огненный, цвета портвейна, кроваво-красный, кавернозный, сосудистый, бородавчатый)
- Q82.8 Другие уточненные врожденные аномалии кожи
Аномальные ладонные складки
Добавочные кожные метки
Доброкачественная семейная пузырчатка (болезнь Хейли-Хейли)
Кожа вялая [cutis-lаха](гиперэластическая)
Дерматоглифические аномалии
Наследственный кератоз ладоней и стоп
Кератоз фолликулярный [Дарье-Чайта]
· Q82.9 Врожденная аномалия развития кожи неуточненная
Q84 Другие врожденные аномалии [пороки развития] наружных покровов
- Q84.0 Врожденная алопеция
- Q84.1 Врожденные морфологические нарушения волос, не классифицированные в других рубриках
o Бусовидные волосы
o Узловатые волосы
o Кольцевидные волосы
- Q84.2 Другие врожденные аномалии волос
- Гипертрихоз
- Сохранившиеся пушковые волосы
- Q84.3 Анонихия
- Q84.4 Врожденная лейконихия
- Q84.5 Увеличенные и гипертрофированные ногти
- Врождённое выпадение ногтей
- Пахионихия
- Q84.6 Другие врожденные аномалии ногтей
- Булавовидные ногти
- Врождённая койлонихия
- Q84.8 Другие врожденные аномалии наружных покровов
- Врождённая аплазия кожи
- Q84.9 Порок развития наружных покровов неуточненный
Q85 Факоматозы, не классифицированные в других рубриках
- Q85.0 Нейрофиброматоз незлокачественный
- Q85.1 Туберозный склероз
- Болезнь Бурневилля
- Эпилойя
- Q85.8 Другие факоматозы, не классифицированные в других рубриках
o Синдромы:
- Пейтца-Егерса
- Старджа-Вебера
- Гиппеля-Линдау
- Q85.9 Факоматоз неуточненный
Рассмотрим некоторые из указанных в МКБ пороков развития кожи и её придатков.
Врожденный ихтиоз
Ichthyosis соngеnitа передается аутосомно-рецессивно с частотой 1:300000 и характеризуется нарушением рогообразования. На коже образуется наслоение плотно спаянных с подлежащими слоями эпидермиса роговых масс, трудно снимаемых при поскабливании. У части больных имеются деформации и уродства в виде расщелины верхней губы, челюсти, твердого и мягкого неба, неполного раскрытия глазных щелей, деформацией ушных раковин, наличия шестого пальца, отсутствие ногтей и волос. Течение болезни хроническое, монотонное без склонности к улучшению. Сочетание кератита, приводящего к потере зрения, ихтиоза, глухоты и других пороков развития описывается в литературе под названием КИД (КID)-синдрома (от первых букв слов: Кеrаtitis. Ichthyosis, Dеаfnеss). При этом синдроме имеются врожденный дефект нервной системы и дефицит клеточного иммунитета.
Больные врожденным ихтиозом уже с раннего детства становятся инвалидами и требуют постоянной посторонней помощи для обеспечения их жизнедеятельности. Они рано осознают свое состояние, считая себя неполноценными людьми, что приводит в дальнейшем нередко к невротическим, ипохондрическим состояниям с возможностью суицидных попыток. Существует несколько клинических форм врожденного ихтиоза, главные из которых - это врожденный пластинчатый ихтиоз и врожденная ихтиозиформная эритродермия (сухая и буллезная формы). В зависимости от времени развития и тяжести течения врожденного пластинчатого ихтиоза различают очень тяжелую (фетальную) и облегченную формы.
При фетальном типе врожденного пластинчатого ихтиоза иногда могут быть роды мертвым плодом, в других случаях рождаются недоношенные, резко ослабленные дети, иногда с отсутствием глотательного рефлекса, затруднением дыхания. Поражение кожного покрова универсальное в виде эритродермии. Ребенок родится как бы покрытый коллоидной пленкой (Соllоdium Ваbу, плод Арлекина),

состоящий из массивных роговых наслоений, между которыми имеются глубокие, часто кровоточащие трещины. Ушные раковины деформированы, веки вывернуты, нос сплющен, рот открыт и напоминает “рыбий рот”. Иногда веки и ушные раковины отсутствуют. Рот, нос и уши заполнены мощными наслоениями роговых чешуйко-корок, которые после удаления быстро возникают вновь. Между пальцами рук и ног иногда имеются перемычки. Кожа лица натянута, мимика отсутствует. Вокруг рта появляются глубокие радиальные трещины. Конечности отечны, почти неподвижны, покрыты наслоениями роговых масс. Такие дети, как правило, погибают в первые часы или дни жизни.
Облегченная форма врожденного пластинчатого ихтиоза совместима с жизнью. Проявления болезни имеются уже при рождении, хотя у некоторых детей они могут появиться после рождения. Состояние больных при рождении тяжелое. Поражение кожи универсальное в виде эритродермии, более выраженное в складках. На всей поверхности кожи отмечается наслоение массивных пластинчатых пергаментообразных чешуек, плотно сцепленных с подлежащими слоями эпидермиса и снимающихся с трудом. Лицо маскообразно, отмечаются выворот век, деформация ушных раковин, кератодермия ладоней и подошв, утолщение кожи крупных складок, в которых легко возникают кровоточащие трещины, могут отсутствовать волосы и ногти.

Такие дети ослаблены, плохо развиваются, не прибавляют массу тела. Иногда они истощены, анемизированы. Часто присоединяются заболевания верхних дыхательных путей, пневмонии, отиты, кандидозные и стафилококковые поражения кожи. Могут наблюдаться кератоконъюнктивиты с помутнением роговицы, пигментный ретинит, глаукома, страбизм, астигматизм, остеопороз, арахнодактилия, конская стопа, синдактилизм, остеолиз, микроцефалия. В патогенезе заболевания основное значение имеет врожденный иммунодефицит как клеточного, так и гуморального звена, а также дефицит витаминов А, Е.
Разновидностью врожденного ихтиоза является врожденная ихтиозиформная эритродермия Брока, описанная в 1902 г. При этой форме наиболее поражены крупные складки. Различают сухой и буллезный типы болезни.
Сухой тип врожденной ихтиозиформной эритродермии клинически мало отличается от проявлений облегченного типа пластинчатого врожденного ихтиоза. Однако при сухом типе всегда наблюдаются очень выраженное поражение кожи крупных складок, шеи, тыла кистей и стоп и более массивные наслоения роговых масс на ладонях и подошвах.
Буллезный тип ихтиозиформной эритродермии описан П. В. Никольским в 1899 г. под названием врожденного универсального акантокератолиза. Эту форму болезни называют в настоящее время также эпидермолитическим гиперкератозом и рассматривают как сочетание ихтиоза с буллезным эпидермолизом. Заболевание наследуется аутосомно-доминантно. При нем были установлены нарушения в системе НLА в локусе АW31. Дерматоз выявляется у ребенка уже при рождении. Кожа гиперемирована, покрыта наслоениями роговых чешуек линейной или ромбовидной формы, что больше всего выражено в складках, на тыле кистей и стоп, шее, где иногда образуются трещины. Роговые чешуйки имеют грязно-серую с зеленоватым оттенком окраску. Наряду с указанными явлениями гиперкератоза, наблюдаются пузыри. После разрушенных крупных пузырей из-за наличия эрозий и ярко гиперемированной кожи вокруг ребенок имеет вид “ошпаренного”. Высыпание пузырей всегда сопровождается нарушением общего состояния ребенка и повышением температуры. В области ладоней и подошв отмечается кератодермия. Наблюдается усиленный рост ногтей и волос. Других деформаций и уродств, как правило, не бывает.
Кроме того, существуют редкие разновидности врожденного ихтиоза, называемые синдромами Рефсума, Руда, Шегрена-Ларссона, Нетертона.
Для синдрома Рефсума характерны ихтиозиформная эритродермия в сочетании с поражением глаз в виде пигментного ретинита с гемералопией, хронической поли-невропатией, проявляющейся прогрессирующими парезами дистальных частей тела, ослаблением или отсутствием рефлексов, повышением количества спинно-мозговой жидкости с нормальным цитозом, симметричная дисплазия суставов, различные изменения ЭКГ.
При синдроме Руда отмечаются врожденная ихтиозиформная эритродермия, тотальная алопеция, инфантилизм, карликовость, отставание полового развития, нервно-психические расстройства в виде умственной отсталости, идиотии, эпилепсии, полиневрита, мышечной атрофии, пигментный ретинит, анемии пернициозоподобного типа.
Синдром Шегрена-Ларссона характеризуется врожденной ихтиозиформной эритродермией, спастическими параличами нижних конечностей, дегенерацией сетчатки глаз, значительным ослаблением зрения, олигофренией.
Синдром Нетертона проявляется врожденной ихтиозиформной эритродермией, сухостью и ломкостью волос, бровей и ресниц (при микроскопическом исследовании выявляются узловатые утолщения, в результате чего волос напоминает бамбук). Часто развивается крапивница или ангионевротический отек.
Диагноз. Распознавание врожденного ихтиоза нередко вызывает затруднения среди как педиатров, так и дерматологов. Правильная диагностика основывается на наличии всех проявлений болезни уже при рождении ребенка или вскоре после него и на явлениях эритродермии с наслоением трудно снимающихся роговых пластинок. Дифференциальный диагноз проводят с десквамативной эритродермпей Лейнера, эксфолиативным дерматитом Риттера, врожденным сифилисом (диффузная папулезная инфильтрация), врожденным буллезным эпидермолизом.
Лечение оставалось долгое время малоперспективным.С 1965 г. в кожной клинике Ленинградского педиатрического медицинского института под руководством Л. А. Штейнлухта началась разработка методики комплексного лечения врожденного ихтиоза у новорожденных детей, которая в дальнейшем продолжала нами совершенствоваться. В настоящее время эта терапия сводится к следующему.
После установления в родильном доме диагноза, в зависимости от тяжести общего состояния ребенка, степени выраженности изменений на коже, исходя из его жизненных потенций, назначаются сразу же с первых дней жизни, один из глюкокортикоидных гормонов. Преднизолон, урбазон, триамцинолон (полькортолон, кеналог) дают по 1,5-3 мг/кг массы тела в сутки (в расчете на преднизолон). Суточная доза препарата дается в два приема: утром (в 8 ч) и днем (в 14-15 ч) после кормления (в утренний прием вводят 2/3 дозы препарата). Детям, у которых затруднено глотание или оно невозможно, препарат дают парентерально. Назначают также препараты калия - 5% хлорид или 5% ацетат по 1/2 чайной ложки 3 раза в сутки (в молоке) и анаболические стероиды (неробол, ретаболил).
Продолжительность введения максимальной дозы глюкокортикоидных препаратов - 1-1,5 мес и зависит от тяжести заболевания, общего состояния ребенка, динамики течения кожного процесса, кинических и биохимических показателей крови (общий белок, белковые фракции, сахар крови, протромбин, электролиты) и мочи. Снижение дозы глюкокортикоидов проводится по 1 мг через 3-5 сут в продолжение 3-4-6 нед. Общий срок терапии больных врожденных ихтиозом составляет около 50 дней, а при тяжелых формах - 70-80 и даже 100 дней.
В комплекс лечения входят внутривенные вливания белковых препаратов: 10% раствора альбумина, нативной свежей плазмы (проверенной на уровень трансаминаз). При выраженной анемии вводят консервированную кровь из расчета 8-10 мл/кг массы. Вливание белковых препаратов следует чередовать с внутривенным введением 5% раствора глюкозы, солевых растворов с добавлением 1 мл 5% раствора аскорбиновой кислоты, 25 мг кокарбоксилазы. Вливание белковых препаратов и растворов проводят через 1 день. В нос (в каждую ноздрю) закапывают по 1 капле 3,44% масляного раствора ретинола ацетата, а при наличии эктропиона - по 1 капле в глазную щель. Внутримышечно вводят 1% раствор пиридоксина по 0,2-0,3 мл и по 15 мкг витамина В12 через 2 дня.
Кормящим грудью матерям дают раствор ретинола ацетата по 50 000 МЕ 1 раз в сутки во время еды, а также назначают внутрь поливитамины.
Новорожденные, страдающие врожденным ихтиозом, требуют особенно тщательного ухода. Их следует содержать в кувезе, в котором поддерживается постоянная температура 36-37 °С. При нарушении глотания кормление проводят через зонд. Белье должно быть стерильным. Из-за пониженной защитной функции кожи таким детям ежедневно делают ванны с добавлением в воду калия перманганата 1:15000 (избегать переохлаждения!). Кожу обрабатывают смягчающими кремами (“Спермацетовым”, “Ланолиновым” с добавлением к ним масляных растворов витаминов А и Е), растительными маслами (персиковым, абрикосовым, оливковым). При мацерации складок применяют 1% водные растворы анилиновых красителей, мази с содержанием 2% нафталана и ихтиола или мази с глюкокортикоидными гормонами.
В случае необходимости (присоединение пиококковой инфекции кожи, отита, пневмонии, пиелонефрита) ребенку назначают антибактериальную терапию (вводят парентерально антибиотики). Целесообразно применение обычного (противокоревого) гамма-глобулина по 1/2 человеко-дозы на инъекцию через 2 сут. Всего вводят 5-6 человеко-доз. Назначают также масляный раствор витамина Е по 100 мг в сутки.
Необходимо еще раз подчеркнуть, что глюкокортикоидная терапия новорожденных детей, страдающих врожденным ихтиозом, эффективна лишь при назначении ее с первых дней, а лучше даже с первых часов жизни больного ребенка. Начатое же после 12-14 дней жизни лечение по данной схеме практически безрезультатно. Поэтому детям более старшего возраста, страдающим врожденным ихтиозом, лечение глюкокортикоидными гормонами противопоказано, так как не только не дает эффекта, но и небезразлично для развивающегося организма ребенка и даже опасно из-за возможных побочных действий этих препаратов.
Лечение более старших детей должно, следовательно, проводиться назначением витаминов А и Е длительными циклами, гигиенических ванн, смягчающих кремов, отшелушивающих мазей. Эрозированные участки кожи обрабатывают алазолем - аэрозолем, содержащим масло облепихи и левомицетин.
В последние годы у двух больных мы наблюдали весьма положительный эффект от применения внутрь ароматического ретиноида - тигазона. Поскольку прекращение приема препарата вызывало рецидив заболевания, необходимо перманентное использование тигазона в поддерживающих дозах.
Прогноз зависит от степени тяжести заболевания и своевременности начала лечения глюкокортикоидными гормонами. У большинства больных, получавших эту терапию с первых дней и часов жизни, проявления врожденного ихтиоза разрешились, умственное и физическое развитие детей не отличалось от сверстников при длительных сроках наблюдения (от 10 до 20 лет).
Врожденный буллезный эпидермолиз
Различают простые и дистрофические формы этого дерматоза.
Простой врожденный буллезный эпидермолиз описан в 1886 г. Кебнером. Характеризуется аутосомно-доминантным типом наследования с частотен 1:50 000. Имеются наблюдения о передаче этого дерматоза в 8 поколениях подряд.
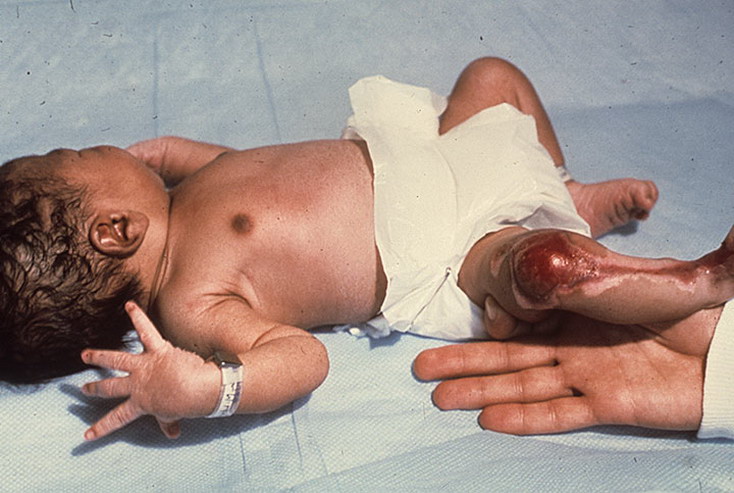
Заболевание начинается с рождения либо вскоре после него. На местах, подвергающихся механическому раздражению (трение, давление, ушиб), возникают тонкостенные пузыри величиной от горошины до грецкого ореха, с серозным содержимым. Воспалительные изменения в окружности пузырей отсутствуют. Располагаются они в области коленных, локтевых суставов, лодыжек, кистей, волосистой части головы.
Для новорожденных с любой разновидностью врожденного апидермолиза типично, что уже сам акт родов оказывается той первой механической травмой, которая приводит к появлению у ребенка пузырей на местах, плотно соприкасавшихся с родовыми путями матери (голова, конечности, ягодицы и др.).
После самостоятельного (или искусственного) вскрытия пузырей происходит быстрое заживление образовавшихся эрозий без атрофии и рубцов, но с временной пигментацией. Симптом Никольского отрицателен. Очень редко (у 2-3% больных) отмечается поражение слизистых оболочек. Ногтевые пластинки не изменяются. У 25% больных выявляется ладонно-подошвенный гипергидроз. Общее состояние больных не нарушено. Течение заболевания легкое, особенно у девочек. Умственное и физическое развитие ребенка не отстает. Обострение заболевания наблюдается в периоды, когда ребенок начинает ползать, а затем - ходить, а также после теплых ванн и в летнее время года. Ремиссии наступают зимой и учащаются к пубертатному периоду.

Локализованная, или летняя, форма простого эпидермолиза (синдром Вебера-Коккейна) начинается на 1-м или 2-м году жизни, иногда в юношеском возрасте, передается по аутосомно-Доминантному типу. Пузыри локализуются исключительно на ладонях и подошвах, покрышки их более толстые, содержимое серозное, нередко серозно-геморрагическое. Часто выявляется гипергидроз ладоней и подошв. Обострения наблюдаются только в жаркое время года, после теплых ванн, при ношении теплой обуви.
Дистрофический врожденный буллезный эпидермолиз
Гиперпластическая разновидность передается по аутосомно-доминантному типу. Болезнь отмечается с рождения или спустя несколько дней. Пузыри изредка встречаются спонтанно, а обычно возникают при незначительном механическом раздражении. Например, даже при ударе самим ребёнком игрушкой-побрякушкой несколько раз по одному и тому же участку (колени, стопы, кисти, живот) через несколько часов появляется гиперемия, а затем пузыри с серозным или серозно-геморрагическпм содержимым, после заживления которых может оставаться рубцовая атрофия. Симптом Никольского отрицательный. На слизистых оболочках часто видны лейкоплакии. Общее состояние не нарушается, умственное и физическое развитие не страдает. Волосы и зубы не изменяются. У некоторых детей отмечаются сухость кожи, гиперкератоз и гипергидроз ладоней и подошв.
Прогноз благоприятный, течение заболевания значительно улучшается при наступлении пубертатного периода.
Полидиспластическая разновидность дистрофического буллезного эпидермолиза наследуется по аутосомно-рецессивному типу и является одной из наиболее тяжелых форм среди всех буллезных эпидермолизов. Заболевание начинается с рождения в виде распространенных пузырей па коже и слизистых оболочках, которые в большом количестве могут появляться не только после травмы, но и спонтанно. Они имеют серозное, но чаще геморрагическое содержимое. После вскрытия образуются медленно заживающие эрозии и язвы. Поражения локализуются не только на конечностях, но и на других местах, захватывая иногда значительные поверхности (спину, грудь, живот, конечности). Симптом Никольского положителен. Часто отмечается значительный зуд. Ногтевые пластинки очень быстро атрофируются и вовсе исчезают. Повторное образование пузырей и язв с последующим их рубцеванием приводит к образованию контрактур, а иногда и мутиляций, когда вместо кистей или стоп остаются обезображенные культи. На рубцах видны эпителиальные кисты (милиумы).
Нередко наблюдаются различные дисплазии в виде ксеродермии, гипотрихоза, акроцианоза, эпдокринопатий, аномалий зубов (которые подвержены быстрому кариесу). Часто отмечаются тяжелые поражения слизистых оболочек полости рта, рубцовое укорочение уздечки языка, поражения гортани, бронхов, пищевода, заднего прохода, приводящие к стенозированию, рубцеванию, а иногда к перфорациям.
Могут возникнуть язвы тонкой и толстой кишки, желчного пузыря, стенозирование мочевыводящих путей, вызывающее задержку мочи, гипертрофию мочевого пузыря и гипернефроз. Возможны сращения конъюнктивы (синблефарон), эрозии роговой оболочки глаз.
Общее состояние больных тяжелое, упитанность иногда резко понижена, наблюдаются малокровие, значительное отставание физического, а у некоторых детей и психического развития. Снижена резистентность к инфекциям (любая разновидность буллезного эпидермолиза часто осложняется вторичной пиококковой инфекцией). Иногда развивается пиелонефрит или гломерулонефрит. Самым тяжелым осложнением является вторичный амилоидоз с преимущественным поражением почек, приводящий к инвалидизации и в итоге - к летальному исходу - в пубертатном периоде, либо после него.
Злокачественная (летальная) форма врожденного буллёзного эпидермолиза была описана датским педиатром Герлицем в 1935 г. Передается аутосомно-рецессивно и проявляется при рождении в виде распространенных геморрагических пузырей на коже, слизистые оболочках рта, гениталий, в области трахеи, бронхов пищевода, желудка и кишечника. Пузыри быстро сливаются между собой, а после их вскрытия остаются на коже и слизистых оболочках болезненные, плохо заживающие, кровоточащие язвенно-некротические очаги. Симптом Никольского положителен. Рубцов и милиа не бывает. Отмечаются врожденные дистрофии ногтей, атрофические изменения скелета. Течение заболевания очень тяжелое, быстро осложняется пиококковой инфекцией, сепсисом и заканчивается летально в первые месяцы жизни ребенка.
С целью пренатальной диагностики генодерматозов в последние годы разрабатываются методы фетоскопической биопсии кожи с последующим световым, и особенно ультрамикроскопическим, исследованием. Биохимическим маркером рецессивного дистрофического буллезного эпидермолиза является увеличение активности в биоптатах кожи плода коллагеназы - в 3,5 раза выше, чем в здоровых клетках.
Патогенез врожденного буллезного эпидермолиза изучен недостаточно. Определенную роль в развитии его отводят возникновению десмосомальных дефектов в дерме, повышенному синтезу и высокой активности коллагеназы, что приводит к разрушению коллагена; при механической травме она активируется из своего профермента. Изменения коллагеназы носят генетический характер.. Кроме того, в сыворотке крови больных выявляется повышенный уровень протеолитического фермента - химотрипсина, который, нарушая целостность базальной мембраны, способствует образованию пузырей. Считается, что химотрипсин может выделяться клетками Лангерганса, или лимфоцитами и кератиноцитами. При буллезном эпидермолизе типа Вебера-Коккейна обнаружено снижение клеточного иммунитета при количественно неизменном гуморальном иммунитете.
Лечение из-за отсутствия специфических методов является весьма затруднительным. Основное внимание уделяют профилактике травм и предотвращению вторичной инфекции. В периоде новорожденности и раннего детства, когда иммунные механизмы еще недостаточно зрелые, особенно важны предупреждение развития сепсиса и быстрое адекватное лечение его. Необходимо грудное вскармливание ребенка.
При тяжелых формах буллезного эпидермолиза наиболее эффективно предупреждают высыпание пузырей глюкокортикоидные гормоны. Назначение кортикостероидов может спасти жизнь детей, страдающих летальной разновидностью болезни. Таким детям вначале применяется преднизолон - до 40-60 мг в сутки, после улучшения состояния дозу препарата постепенно снижают вплоть до полной его отмены. Назначают также анаболические средства (неробол, нероболил), парентеральное введение антибиотиков широкого спектра действия, вливание альбумина, плазмы.
При обострениях полидиспластической и гиперпластической форм эпидермолиза целесообразны лишь короткие курсы глюкокортикоидных гормонов из расчета 1-2 мг/кг (в расчете на преднизолон) внутрь в течение 10-15 дней с последующим постепенным снижением дозы вплоть до полной отмены препарата. Использование относительно небольших доз преднизолона эффективно в профилактике возникновения пузырей на слизистых оболочках, стриктур пищевода, образования синдактилий, глазных и зубных осложнений.
Необходим систематический контроль за состоянием крови с целью своевременного выявления и лечения анемии. Для профилактики образования контрактур важно обеспечить полный объем движений во всех суставах. С целью устранения гиперплазии десен следует приучить ребенка регулярно полоскать полость рта.
У многих больных, особенно при гиперпластической форме эпидермолиза, благоприятный эффект наблюдается при назначении повторных курсов (по 2-3 мес с перерывом в 2-3 нед) токоферола ацетата, восстанавливающего нормальную структуру коллагеновых и эластических волокон дермы. С целью иммунокорригирующего и общеукрепляющего воздействия назначают нуклеинат натрия, метацил, вливания плазмы, гемодеза, повторные курсы по 10-12 инъекций гамма-глобулина, витамины А, С, В1, D, рутин, никотиновую кислоту, пиридоксин, витамин В12, кальция пантотенат и пангамат, препараты железа. А. В. Смирновым (1986) получено улучшение в клиническом течении полидиспластической формы буллезного эпидермолиза у двух детей (2 года и 6 лет) при внутримышечном введении солкосерила по 2 мл ежедневно в течение 10 дней.
При дистрофических формах болезни изучают препарат цианидалон-3, являющийся производным флавонов и относящийся к стабилизаторам мембран. При использовании его отмечалось более спокойное течение заболевания, уменьшалось образование пузырей, лучше заживали эрозии, что препятствовало появлению стриктур пищевода, мутиляцпй и синдактилий. Этот препарат in vitro повышает устойчивость коллагена против воздействия коллагеназы.
Наружная терапия: возникающие пузыри необходимо вскрывать в двух местах стерильной иглой, смазывать покрышки 1-2% водным раствором анилинового красителя (фуксин, эозин, бриллиантовый зеленый, метиленоный синий). Используют примочки с чаем, корочки смазывают оливковым маслом. На эрозии и язвы регулярно применяют ируксоловую мазь, мазь солкосерила, дерматоловую (3-5%), гелиомициновую мази, наносят масляные растворы витаминов А, Е, D, смешанных в равных пропорциях. Назначают также мази, кремы и аэрозоли, содержащие глюкокортикоидные гормоны, антибиотики, облепиху. Следует по возможности избегать применения повязок и наклеивания пластыря, для предупреждения трения, травмирования кожи. Хороший эффект оказывают общие лечебные ванны с отварами дубовой коры, калины, ромашки, череды, а также с раствором калия перманганата, цинка сульфата и ванны с добавлением оливкового масла.
Эффективно общее УФО: снижая активность клеток Лангерганса, оно уменьшает продукцию ими химотрипсина (уровень его в плазме крови значительно снижается), что препятствует образованию пузырей.
При мытье ребенка необходимо избегать механического травмирования кожи. Для одежды и обуви следует употреблять мягкие материалы, избегать горячей и твердой пищи. В настоящее время возможна успешная ортопедическая реконструкция искалеченных кистей.
Дети, страдающие буллезным эпидермолизом. особенно тяжелыми формами, должны находиться под диспансерным наблюдением дерматолога, педиатра, ортопеда и периодически получать комплексное лечение в стационаре.
Первичная (врожденная) лимфедема.
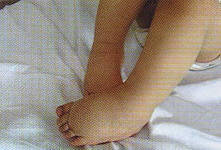
Обусловлена врожденной патологией лимфатических сосудов. Как правило, развивается в детстве и юности (80% больных – девочки в возрасте до 18 лет). Чаще поражает нижние конечности, реже – верхние. Обычно бывает двусторонней. В 6% случаев врожденная лимфедема вызвана наследственным заболеванием (синдром Нонне-Милроя, синдром Мейжа). У остальных 94% больных первичная лимфедема развивается из-за врожденной аплазии или гипоплазии лимфатических сосудов. Компенсированная первичная лимфедема нередко быстро прогрессирует после беременности или травмы. Врожденная лимфедема вначале поражает дистальные отделы конечностей (стопы или кисти). У пациентов появляется безболезненный плотный отек пальцев, распространяющийся на стопу и голеностопный сустав (при поражении верхней конечности – на кисть и лучезапястный сустав). По мере прогрессирования лимфедемы отеки распространяются на голень и бедро. Ноги больного становятся похожими на колонны. В области суставов со временем образуются складки из отечных мягких тканей. Складки на тыле стопы не выражены. Кожа напоминает апельсиновую корку.Пациенты с лимфедемой наблюдаются флебологами и сосудистыми хирургами.
Ксеродерма пигментная
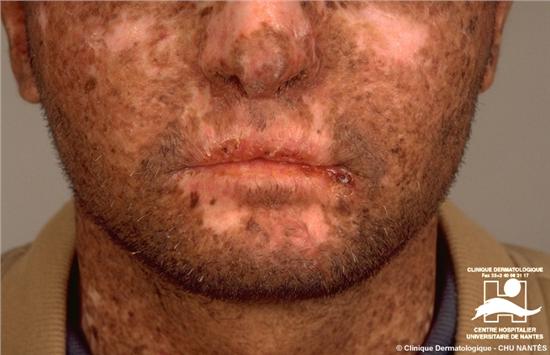
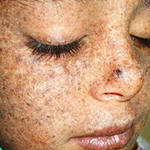
Ксероде́рма пигме́нтная (xeroderma pigmentosum; греч. xeros сухой + derma кожа; синоним: ретикулярный прогрессирующий меланоз, Пика прогрессирующий ретикулярный меланоз)-наследственное заболевание кожи, характеризующееся ее повышенной чувствительностью к ультрафиолетовому излучению; является облигатным предраком кожи. Встречается редко, наследуется по аутосомно-доминантному типу. Генетический дефект заключается в отсутствии или малой активности эндо- и экзонуклеаз-полимераз, устраняющих повреждения ДНК клеток кожи, вызванные УФ-излучением. При К. п. известны два вида нарушений генотипа: недостаточность фермента УФ-эндонуклеазы, вычленяющего поврежденные УФ-излучением участки ДНК, и дефект фермента ДНК-полимеразы, участвующего в воссоединении разрывов ДНК. В последнем случае у больных отмечается также повышенная чувствительность кожи к ионизирующему излучению. Такой ферментативный дефект приводит к нарушению пигментации кожи, ороговения, к атрофическим изменениям эпидермиса и дистрофии соединительной ткани, в заключительной стадии — к клеточной атипии и злокачественному росту.
В клинической картине заболевания независимо от генотипа выделяют три стадии. Первая стадия проявляется обычно на 2—3-м году жизни (редко позже) в весенне-летний период, после пребывания на солнце. Отмечается стойкая воспалительная реакция кожи на открытых участках тела (лицо, шея, кисти, предплечья). Она характеризуется эритемой, шелушением кожи с последующим развитием неравномерной гиперпигментации по типу лентиго, веснушек . Каждое последующее пребывание на солнце усиливает эти процессы. Через несколько лет отчетливо выявляется вторая стадия, при которой участки атрофии кожи различных очертаний и величины, телеангиэктазии в сочетании с неравномерной пигментацией кожи придают ей пестрый вид -пойкилодермия, напоминающий клиническую картину хронического радиационного дерматита . На отдельных участках кожи могут образовываться бородавчатые разрастания, трещины, корки, изъязвления, происходит экзематизация. Атрофические изменения кожи лица сопровождаются истончением хрящей носа, ушных раковин, деформацией естественных отверстий (сужение носовых ходов, ротового отверстия), выворотом век,
нарушением их роста, помутнением роговицы, слезоточивостью и светобоязнью. В третьей стадии болезни, которая развивается обычно в юношеском возрасте, иногда значительно раньше, в очагах поражения образуются доброкачественные и злокачественные опухоли (фибромы, ангиомы, кератомы, базалиомы, меланомы). Особенно выражена тенденция к малигнизации с метастазированием во внутренние органы бородавчатых очагов поражения. В результате этого 2/3 больных погибают в возрасте до 15 лет.
Наиболее тяжелой формой заболевания, которая наследуется по рецессивному, сцепленному с Х-хромосомой типу, является ксеродермическая идиотия (синдром Де Санктиса — Каккьне). При этой форме К. п. выражены нарушения со стороны ц.н.с., недостаточное развитие гипофиза и мозжечка, микроцефалия. Отмечаются идиотия, парезы, судороги, координационные и рефлекторные расстройства. Кроме того, характерны задержка роста, полового развития, наблюдается потеря слуха, нарушение порфиринового обмена.
Диагноз основывается, как правило, на данных клинической картины (связь процесса с солнечным облучением, поражение открытых участков кожи по типу пойкилодермии с последующей малигнизацией). В начальном периоде заболевание дифференцируют с фотодерматозами ,пойкилодермией.
Лечение на ранней стадии заболевания проводится амбулаторно дерматологом. Назначают синтетические антималярийные препараты (например, хингамин), которые противодействуют деполимеризации ДНК, уменьшают чувствительность кожи к солнечным лучам; витамины А, РР, группы В; наружно рекомендуются кортикостероидные мази, на бородавчатые разрастания — цитостатические. Применяют также фотозащитные средства с SPF 50+ . При развитии опухолей больного необходимо направить к онкологу. При ксеродермической идиотии лечение проводится невропатологом в специализированном стационаре.
Прогноз неблагоприятный ввиду выраженной склонности процесса к малигнизации. Первичная профилактика не разработана. В целях замедления прогрессирования процесса больные должны находиться под диспансерным наблюдением дерматолога, онколога и при необходимости офтальмолога и невропатолога. Важны мероприятия по защите от солнечных лучей (больным следует носить широкополые шляпы, перчатки, пользоваться зонтиками, ходить по теневой стороне улицы). При появлении бородавчатых разрастаний целесообразно их оперативное удаление в ранние сроки во избежание малигнизации.
Мастоцитоз
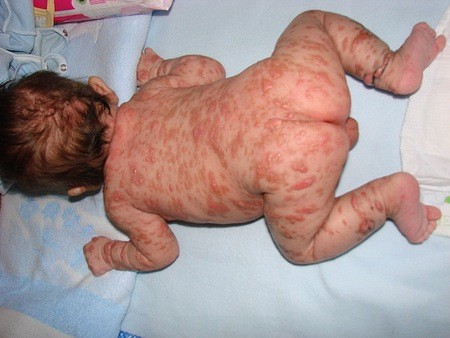
Первые проявления мастоцитоза возникают у 70% детей в возрасте от 1 до 12 мес, но могут болеть и новорожденные, а также дети более старшего возраста и взрослые лица. Заболевание имеет стадийность: прогрессирующая стадия, стабилизация и регрессирующая стадия. Для прогрессирующей стадии характерны приступообразные высыпания пятен, папул, волдырей розово-красного цвета, округлой или овальной формы. Размер их от просяного зерна до 2- 3 см в поперечнике, иногда они имеют вид более крупных бляшек. Локализация сыпи - туловище, конечности, волосистая часть головы, иногда лицо, редко ладони и подошвы. Слизистые оболочки не поражаются. Ребенок нередко беспокоен из-за зуда.
Патогномоничным для мастоцитоза считается симптом Унны-Дарье, когда при трении шпателем или пальцем пятна или папулы или после прикосновения к ним теплого предмета вскоре появляются покраснение и набухание этого элемента - он приобретает волдыреобразный вид.
Набухание и отечность элементов сыпи, иногда весьма значительное, наблюдается после мытья ребенка в теплой, ванне, особенно при пользовании мочалкой и даже нежной губкой, что сопровождается нередко сильным зудом.
Симптом Унны-Дарье обусловлен тем, что механическое или термическое раздражение вызывает выход из гранул тучных клеток большого количества гистамина, гепарина, серотонина и гиалуроновой кислоты, что вызывает расширение сосудов, усиление порозности их стенок, выход жидкости в окружающие ткани и усиливает зуд. Возможно рефлекторное появление симптома, когда при потираний пальцем одного из элементов появляется отек близлежащих пятен, не подвергавшихся трению. На здоровой коже этот симптом не проявляется.
Высыпания в прогрессирующей стадии могут периодически исчезать и появляться вновь, что в итоге ведет к более темной окраске элементов сыпи вплоть до коричневого цвета и увеличению их числа - от единичных до сотен.
В стадии стабилизации, наступающей на 2-м году жизни и позднее, прекращается возникновение новых элементов сыпи. В этот период так же, как и в прогрессирующей стадии, наблюдается лишь появление отечности существующих элементов после термического и механического раздражения, а также после ультрафиолетового или солнечного облучения.
Регрессирующая стадия начинается в возрасте после 6-7 лет или к периоду полового созревания и характеризуется постепенным побледнением и иногда даже разрешением элементов.
У детей грудного возраста мастоцитоз проявляется более заметным полиморфизмом высыпаний, среди которых наиболее типичными являются пятнистые и пятнисто-папулезные элементы, несколько реже - папулы, узлы и пузыри. Буллезный мастоцитоз обычно наблюдается уже при рождении ребенка в виде изолированного поражения либо сочетается с пятнистой или узловатой формами. Симптом Никольского отрицательный. Нередко бывают солитарные формы мастоцитоза, когда имеются 1-2-3 узла величиной от горошины до 3-5-копеечной монеты, в центре которых периодически появляются пузыри. Эта форма мастоцитоза у детей имеет более благоприятное течение: через несколько месяцев после появления сыпь полностью разрешается.
Иногда происходит слияние папул и узлов с образованием значительных участков диффузной инфильтрации, иногда сплошь покрывающих туловище ребенка, что придает коже вид “стеганого одеяла”. В этих случаях при малейшем механическом или термическом раздражении кожи, а иногда и спонтанно, происходит ухудшение общего состояния ребенка: усиливаются зуд, покраснение и отечность кожи, появляются беспокойство, раздражительность, плаксивость, тахикардия, головная боль, желудочно-кишечные симптомы в виде анорексии, болей в животе, метеоризма, тошноты, диареи, а иногда развивается гистаминовый шок (что мы наблюдали у некоторых детей).
Системный, или кожно-висцералъный, мастоцитоз у детей грудного возраста встречается очень редко, он наблюдается в основном у взрослых. Отмечается резко выраженный зуд, большое количество волдыреобразных пигментных высыпаний. Появляются головная боль и головокружение, артралгии, увеличение печени и селезенки (вплоть до развития цирроза этих органов). Поражение желудочно-кишечного тракта проявляется тошнотой, рвотой, болями в животе, перемежающимися поносами и другими явлениями гастрита, энтерита. Эти симптомы наблюдаются у 25-50% больных. Имеются рентгенологические изменения костей в виде ограниченного или диффузного сочетания остео-склероза и остеопороза. В костном мозге увеличивается число тучных клеток. Наблюдается безболезненное увеличение и уплотнение подчелюстных, заушных, подмышечных, паховых лимфатических узлов. В крови отмечаются анемия, лейкоцитоз или лейкопения, лимфоцитоз, увеличение СОЭ; повышается уровень гистамина в плазме и в моче. При гистологическом исследовании в пораженных висцеральных органах выявляется диффузная инфильтрация тучными клетками. Встречаются злокачественные формы кожно-висцерального мастоцитоза, при котором, одновременно с поражением кожи, костей и внутренних органов, быстро развиваются миелоидная лейкемия и амилоидоз, что приводит к молниеносному течению заболевания и быстрому летальному исходу.
Диагноз мастоцитоза ставится на основании клинической картины поражения кожи, наличия симптома Унны-Дарье, результатов гистологического исследования элементов сыпи. При необходимости проводят рентгенологическое обследование кистей рук, а также изучают функциональное состояние висцеральных органов.
Гистопатология.В биоптате элемента мастоцитоза в верхней части дермы виден состоящий из тучных клеток инфильтрат, иногда захватывающий всю толщу дермы или проникающий в подкожную жировую клетчатку. В базальном слое эпидермиса большое количество пигмента бурого цвета. При буллезной форме мастоцитоза пузыри расположены субэпидермально.
Этиология и патогенез окончательно не выяснены. Господствует мнение, что мастоцитоз детского возраста носит невоидный характер и, возможно, является наследственным заболеванием, передающимся как аутосомно-доминантно, так и аутосомно-рецессивно. Имеется предположение о связи этой болезни с нарушениями гемопоэза. Системный мастоцитоз относят к группе ретикулёзов.
Лечение - симптоматическое. При выраженом зуде и появлении свежих высыпаний следует на 5-7 дней назначить один из антигистаминных препаратов - тавегил, супрастин, диазолин, фенкарол, пипольфен, димедрол, ципрогептадин. Некоторые врачи считают, что в связи со склонностью пигментной крапивницы у детей к спонтанным ремиссиям лечение ее не показано, тем более, что различные терапевтические мероприятия оказывают малозаметное влияние на течение заболевания, с чем мы вполне согласны.
Улучшает течение кожных форм (особенно буллезных) мастоцитоза у детей грудного возраста кромолин-натрий (интал). Его дают внутрь по 100-300 мг в сутки. Детям грудного возраста в течение длительного времени (до 15 мес). Прекращение приема препарата вызывает через 2-3 нед рецидив заболевания, который купируется повторным назначением интала. Также улучшает течение мастоцитоза у детей задитен (кетотифен), назначаемый внутрь в виде сиропа по 0,125 мл/кг массы тела в 2 приема в течение 4-6 нед. Механизм действия обоих препаратов основан на предупреждении выделения гистамина и других медиаторов вследствие стабилизации мембран мастоцитов кожи и желудочно-кишечного тракта, что оказывает тем самым симптоматическое влияние на течение заболеванияПри системных формах мастоцитоза применяют внутрь глюкокортикоидные гормоны и цитостатические препараты, которые лишь сдерживают развитие болезни.
Прогноз в основном благоприятный, так как у большинства детей проявления мастоцитоза с возрастом постепенно разрешаются. Между тем из-за возможности трансформации мастоцитоза у взрослых в ретикулёз необходимо больных детей подвергать диспансерному наблюдению.
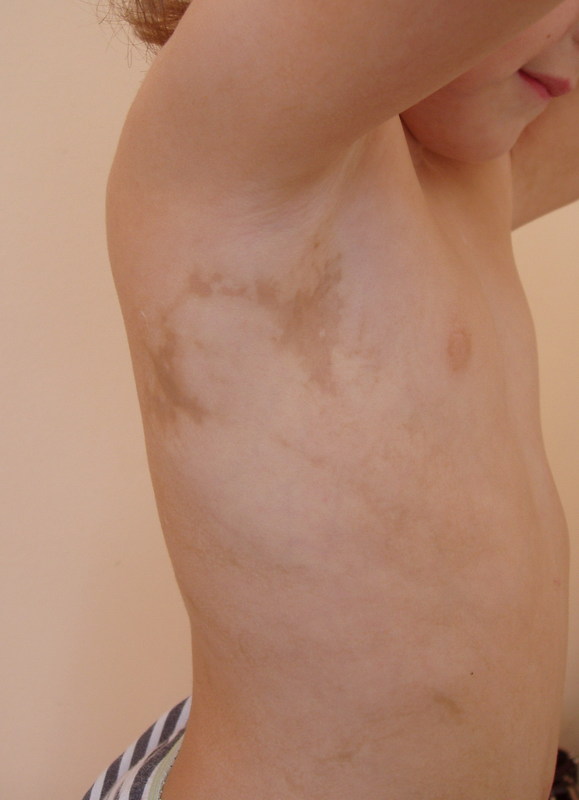
Синдром Блоха-Сульцбергера (недержание пигмента)

Является редким генодерматозом. Имеется ряд описаний этой болезни в нашей стране. Заболевание обнаруживается уже при рождении ребенка или в первые дни и недели жизни.
Постепенно происходит спонтанный регресс элементов сыпи по мере увеличения возраста ребенка, а у взрослых лиц остаточные проявления болезни могут не встречаться.
Болеют обычно только девочки. Отношение больных мальчиков и девочек составляет 6 : 210, частота болезни 1 : 75 000. Признается аутосомно-доминантный тип передачи с манифестацией, ограниченной женским полом и связанной с геном в Х-хромосоме. Этот ген летален для мужских особей, что обусловливает спонтанные аборты у матерей. Имеется предположение о влиянии вирусной инфекции на плод, внутриутробной интоксикации и аллергизации.
Клиника.Характерна пестрая картина элементов сыпи, в результате чего заболевание последовательно проходит 4 стадии развития.
I стадия - эритематозно-везикулезная. Появляются пятна, волдыри, везикулы, пузыри, пустулы, располагающиеся линейно в виде полос вдоль нервных стволов. Локализация элементов сыпи в основном в области боковых поверхностей туловища, на конечностях.
II стадия - гипертрофическая. На инфильтрированном основании появляются лихеноидные, лентикулярные, бородавчатые, гиперкератотические папулы и бляшки, расположенные как симметрично, линейно, так и беспорядочно.
III-пигментная стадия - характеризуется наличием не шелушащихся пятен коричневато-желтого цвета, сравниваемых с брызгами грязи или следами отлива волн.
IV стадия характеризуется участками депигментации и слабо выраженной атрофии. Стадийность элементов сыпи не всегда сохраняется.
Иногда, во 2-й половине 1-го года жизни, на фоне появляющейся пигментации вновь возникают высыпания, свойственные I и II стадиям. Весьма характерна, особенно для I и II стадий заболевания, эозинофилия (40-50% и более) периферической крови. Могут поражаться придатки кожи в виде истончения, обламывания волос, появления зон курчавости, алопеции, изменений ногтей, которые становятся тусклыми, коричневато-бурого или сероватого цвета.
I и II стадии заболевания, существующие, как правило, в первые 2-3 мес жизни, соответствуют синдрому Асбо-Хансена, называемому также “буллезный кератогенный эозинофильный дерматит девочек”.
У некоторых детей клиническая картина болезни ограничивается поражением кожи и ее придатков с постепенным регрессом, вплоть до исчезновения их в период полового созревания. Это характеризует легкое течение заболевания и не требует специального лечения.
Тяжелое течение заключается в системном поражении, прежде всего, нервной системы. Отмечаются микроцефалия, гидроцефалия, менингит, эпилепсия, спастические параличи, олигофрения, задержка физического развития ребенка. Поражаются глаза: увеит, катаракта, кератит, пигментный ретинит, нистагм, птоз, синие склеры, анизокория, страбизм, миопия, отслойка сетчатки, недоразвитие слезопродуцирующего аппарата, воспаление зрительного нерва и его атрофия вплоть до слепоты. Важным признаком, позволяющим установить ретроспективный диагноз в старшем возрасте и у взрослых после исчезновения кожных симптомов, являются аномалии зубов в виде изменения цвета эмали, микродентии, диастемы, отсутствия отдельных зубов. Наблюдаются патологические отклонения со стороны скелета (лишние или недостающие позвонки и ребра, укорочение конечностей), костей черепа. Могут отмечаться нарушения висцеральных органов (аномалии почек, врожденные пороки сердца).
Одной из характерных особенностей болезни у детей грудного возраста, особенно у новорожденных и в первые месяцы жизни, является недостаточность иммунитета, что сопровождается множественными инфекциями респираторных, мочевыделптельных, ЛОР-органов, желудочно-кишечного тракта, центральной нервной системы. Эти инфекции приводят нередко к сепсису и летальному исходу.
Гистопатология.В I стадии - в эпидермисе спонгиоз, наличие эозинофильных гранулоцитов; в дерме - инфильтрат с эозинофилами и мононуклеарами. Во II стадии - акантоз, папилломатоз, гиперкератоз, наличие дискератотических клеток; в дерме - воспалительный инфильтрат, содержащий меланофаги. В III стадии - уменьшение пигмента в клетках базального слоя эпидермиса, которые дистрофически изменены, вакуолизированы. В дерме - в верхней ее части массивные отложения меланина в макрофагах.
Дифференциальный диагноз проводится с пиодермией, врожденным буллезным эпидермолизом,болезнью Дарье, герпетиформным дерматитом, токсидермией, крапивницей, мастоцитозом, пигментным и веррукозным невусами.
Лечение.Специфических методов терапии нет. При наличии пиодермии, сопутствующих инфекций бронхолегочной системы, ЛОР-органов, желудочно-кишечного тракта и других органов проводят антибактериальную терапию антибиотиками широкого спектра действия, применяют инъекции гамма-глобулина, вливания альбумина, плазмы, гемодеза. При тяжелом течении назначают глюкокортикоидные гормоны по 0,5- 1,0 мг/кг массы тела (в расчете на преднизолон) в течение 2-3 нед с последующим постепенным снижением дозы до полной отмены. Целесообразна витаминотерапия: внутрь ретинола ацетат, витамины группы В (В1, В2, В5, В6), аскорбиновая кислота - курсами в течение 1-1,5 мес, внутримышечно аевит по 0,5 мл 2 раза в неделю - 1-1,5 мес. Для профилактики пиодермии элементы сыпи смазывают 3-4 раза в день 1-2% водными или спиртовыми растворами анилиновых красителей (фуксин, эозин, бриллиантовый зеленый, метиленовый синий) и в последующем глюкокортикоидными мазями (преднизолон, фторокорт, флуцинар, лоринден).
Прогноз при легких формах благоприятный, нередко отмечается спонтанное разрешение высыпаний. При тяжелых формах прогноз серьезный, так как заболевание может сопровождаться инвалидизацией с раннего детства и ранним летальным исходом.
Дисплазия эктодермальная ангидротическая (синдром Криста-Сименса-Турена)

Для ангидротической формы эктодермальной дисплазии характерна следующая триада симптомов: гипо- или ангидроз, гипотрихоз, гиподонтия. Кожа истончена, нежноскладчата, особенно вокруг глаз. Потоотделение снижено или реже отсутствует по всму кожному покрову, за исключением мест локализации апокриновых желез, где оно в большей или меньшей степени может сохраняться. Значительна сухость кожи, несколько меньше в подмышечных впадинах, в области половых органов, на лице и слизистых, что часто приводит к развитию конъюктивитов, ларингитов, фарингитов, ринитов, стоматитов. У ряда больных наблюдается ихтиозиформное шелушение, фолликулярный гиперкератоз, ладонно-подошвенные кератодермии. Резкое поредение волос на голове или тотальная алопеция. Брови, а иногда и ресницы редкие, скудное или отсутствует оволосение в подмышечных областях и на лобке. Пушковые волосы обычно сохраняются. Зубы поздно прорезываются, долго сохраняются на стадии молочных, могут быть не в полном количестве, или даже отсутствовать полностью, часто деформированы, с большими промежутками между ними. За счет дистрофии зубов кожа вокруг рта может иметь складки, напоминающие рубцы при врожденном сифилисе.
Характерен внешний вид больных: низкого роста, большой квадратный череп с выступающей лобной частью, массивный подбородок, надбровные дуги, высокие и широкие скуловые кости, впалые щеки, выпуклые губы, большие уши (уши сатира), седловидный нос. Reed и соавт. описали сочетание с acanthosis nigricans.
Из других состояний описана склонность к экзематозным реакциям, атопический дерматит и астма, гипотиреоз, милиумподобные папулезные высыпания, обусловленные сально-железистой гиперплазией, аплазия сосков. У женщин заболевание протекает в смягченной форме в виде нерезко выраженных зубных аномалий, очаговых расстройств потоотделения, слабого развития молочных желез.
Врожденная аплазия кожи

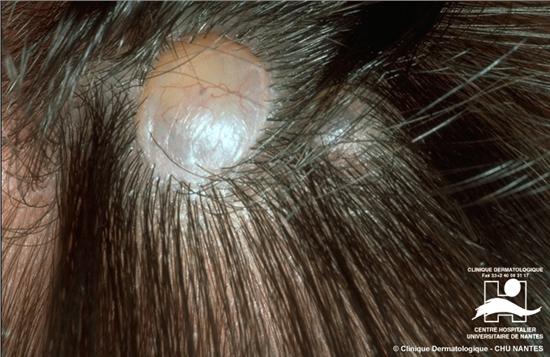
Порок развития, характеризующийся очаговым дефектом кожи. Может сочетаться с аплазией глубже расположенных тканей (в частности, с различными формами дизрафий – незакрытия эмбриональных щелей). аплазия кожи является результатом фетопатии, возникающей при воздействии различных неблагоприятных факторов (вирусная, бактериальная инфекции, интоксикация, внутриматочная или родовая травма и др.). Возможны наследственные формы аплазии с аутосомно-доминантным или рецессивным типом наследования. Описаны случаи хромосомных аномалий (трисомия по 13 паре, делеция короткого плеча 4 хромосомы). Различают несколько видов аплазии кожи в зависимости от наиболее частой ее локализации. врожденная аплазия кожи Андерсона–Нови (синдром Кемпбелла) – аплазия кожи черепа. Характеризуется дефектом кожи (до 10 см в диаметре) мембранозного или буллезного характера, локализующимся обычно в теменной области, реже в затылочной и задней аурикулярной областях. В 70% случаев очаг аплазии одиночный, однако возможно формирование 2-3 очагов поражения, реже более. Мембранозные очаги обычно округлые, лишены кожи, подлежащих тканей (включая костную) и покрыты тонкой эпителиальной мембраной. Гистологически в области мембранозного дефекта обнаруживают компактный коллаген с уменьшением количества эластических волокон, придатки кожи отсутствуют. Возможны признаки воспаления, некроза. Буллезные поражения выявляют обычно у плодов; у новорожденного на месте пузыря сформирован уже язвенный дефект, который в течение нескольких недель заживает с образованием атрофического рубца, в зоне которого волосы не растут (рубцовая алопеция). Врожденная аплазия кожи черепа может сочетаться с врожденной гидроцефалией, гемангиомами, поликистозом почек, менингоцеле, расщеплением неба, микрофтальмией, буллезным эпидермолизом и др. Возможны осложнения в виде присоединения вторичной инфекции, менингита (при сообщении кожного дефекта с корой мозга), кровотечения из сагиттального синуса. врожденная аплазия кожи Девинка – аплазия кожи черепа, туловища или конечностей. Характеризуется участками, лишенными кожи, затянутыми тонкой мембраной, через которую хорошо различимы подлежащие органы и ткани. Возможно сочетание с дефектом развития в виде отсутствия конечностей. ограниченная аплазия кожи Фрейда – аплазия кожи средней линии тела или (симметричная) конечностей. Дефекты кожи носят язвенный характер, медленно заживают с образованием рубца. аплазия кожи Барта характеризуется множественными язвенными дефектами в области родничка, конечностей, слизистых оболочек. В настоящее время рассматривается как вариант буллезного эпидермолиза (см.). Диагноз основывается на клинической картине. Дифференциальный диагноз проводят с механическими повреждениями (во время родов), буллезным эпидермолизом, врожденным сифилисом.
Лечение направлено на предотвращение осложнения процесса бактериальной флорой (при необходимости используют антибиотики); наружно – эпителизирующие и дезинфицирующие средства (1-2% растворы метиленового синего или бриллиантового зеленого, паста Лассара, гиоксизон и др.).
Врожденная алопеция
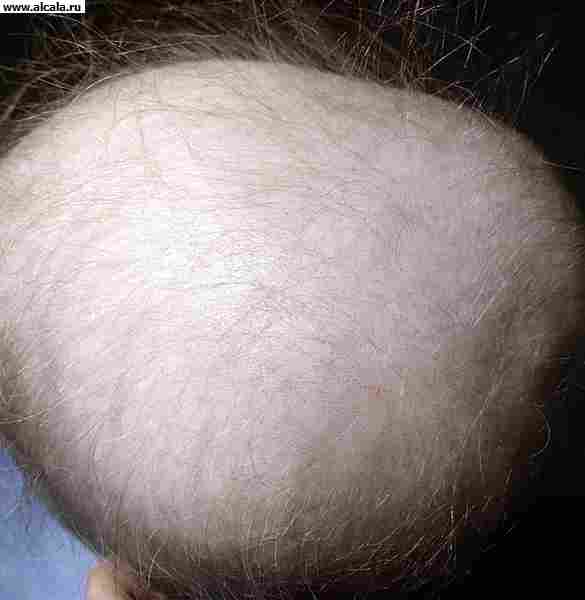
Аlopecia congenita обусловлена дефектом развития, который относят к эктомезодермальным дисплазиям. Врожденная алопеция может быть самостоятельным заболеванием или частью общей патологии, при которой наблюдаются разнообразные дефекты развития ногтей, зубов, костей, эндокринных органов. Эти заболевания нередко имеют семейный или наследственный характер. В их основе лежит частичное или полное отсутствие сально-волосяных фолликулов. Врожденную алопецию могут вызвать генетически обусловленные нарушения синтеза аминокислот, ведущие к нарушению кератинизации волос.
Полное отсутствие волос у детей встречается крайне редко. Значительно чаще наблюдается частичное или резкое уменьшение количества волос. В этих случаях волосы на голове тонкие, короткие, неодинаковой длины, редкие, некоторые из них обломаны. Однако кожа в зонах поредения волос не изменена. Одновременно отмечается поредение бровей: короткие толстые волосы заменяются пушковыми. Ресницы, как правило, отсутствуют или резко разрежены. В подкрыльцовых впадинах и на лобке волосы редкие, изогнутые, короткие. На туловище пушковые волосы нежные, разрежены. В области подбородка и щек количество пушковых волос несколько больше.
Среди врожденных алопеций важное место занимают гипотрихии, развивающиеся у больных, страдающих гипоплазиями и дисплазиями кожи. Кроме изменений кожи, волос, ногтей, зубов, у больных можно выявить различные аномалии развития пальцев, молочных желез. В ряде случаев гипоплазии и дисплазии связаны с невоидными изменениями и появляются не сразу после рождения, а спустя некоторое время, когда выявляется анатомическая или функциональная неполноценность ряда систем, например, эластичной ткани (гиперэластическая кожа), пигментообразования (пигментная ксеродерма), нарушение роста ребенка.
Гипотрихия наблюдается при семейной акрогерии Готтрона, врожденной аплазии кожи, ангидротической эктодермальной дисплазии, синдроме Конради-Хюнерманна, кератодермии Бушке-Фишера, синдроме Поленда (syndromus Poland) - наследственном симптомокомплексе, при котором выявляются аномалии развития груди, плеч, ладоней, сопровождающиеся односторонним отсутствием оволосения в подмышечной впадине.
Гипертрихоз
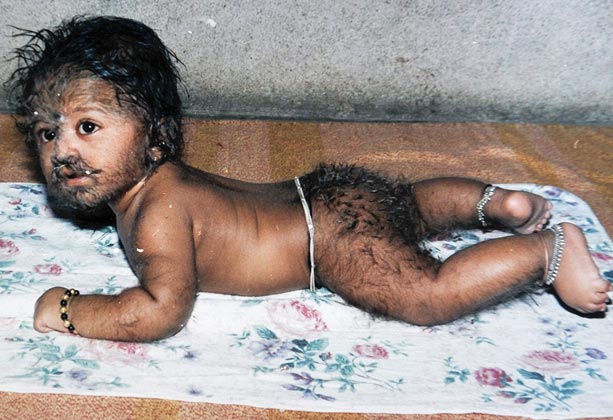
Гипертрихоз или гирсутизм выявляется уже при рождении в виде волосатости всего кожного покрова. По мере развития волосы приобретают шерстеобразный вид и покрывают лицо ребенка, туловище, конечности, за исключением ладоней, подошв, крайней плоти и малых половых губ. Наиболее быстро растут волосы на лице, что придает больному уродливый вид. Таких людей когда-то называли “волосатый человек”, или “человек-собака”. При гипертрихозе дополнительные волосы не вырастают, а наблюдается лишь чрезмерное развитие тех волос, которые в норме являются незаметными, или малозаметным пушком. Одновременно у детей могут наблюдаться аномалии зубов, spina bifidа, осложняющаяся иногда хвостатым отростком, что также обусловлено внутриутробными дефектами развития.
Иногда гипертрихоз имеет ограниченный характер (конечности, лицо). У детей старшего возраста, особенно у девочек, болезнь вызывает тяжелые психические переживания вплоть до возникновения навязчивых идей (трихомания). Заболевание имеет аутосомно -доминантный тип наследования.
Лечение в грудном возрасте не проводится, а в дальнейшей жизни - у подростков и взрослых эффективна фото- и электроэпиляция.
Мониле́трикс

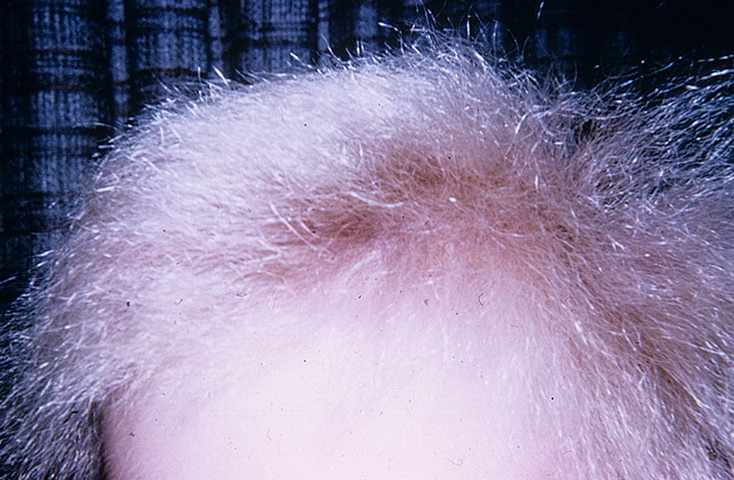
Мonilethrix; лат. monile ожерелье + греч. thrix волос; синоним: четкообразная аплазия волос, веретенообразные волосы, монилиформные волосы-
наследственный кератоз, проявляющийся поражением кожи и волос. Наследуется по аутосомно-доминантному типу. В патогенезе важное значение придается нарушению деятельности эндокринной и нервной систем, которые принимают участие в регуляции питания и роста волос.
Клиническая картина характеризуется появлением на коже волосистой части головы у ребенка первого года жизни мелких узелков с роговыми шипиками на вершине, расположенных около устьев волосяных фолликулов. Постепенно кожа на голове становится сухой, шелушится. Волосы в результате дистрофических изменений становятся сухими, ломкими. лишенными блеска. Их длина не достигает 2—3 см. Они интенсивно выпадают, образуя участки облысения. Возможны также дистрофия ногтей, зубов, фолликулярный кератоз кожи туловища. Иногда М. может сочетаться с катарактой (синдром Сабуро), а также с гипофизарной недостаточностью и сужением полей зрения (синдром Раца-Тури). Течение заболевания хроническое, возможно временное улучшение процесса в период полового созревания.
При микроскопическом исследовании структура волос напоминает ожерелье в связи с чередованием в стержне волоса светлых и темных участков. При этом светлые участки выглядят истонченными из-за отсутствия в них мозгового вещества, а темные (нормальные по структуре) утолщенными.
Лечение проводит дерматолог амбулаторно; назначают витамины А, Е, группы В; показаны УФ-облучение, массаж жидким азотом, втирания 2% серно-салициловой, 5% серно-дегтярной мазей.
Прогноз для жизни благоприятный. Профилактика не разработана.
Папилломатозный порок развития
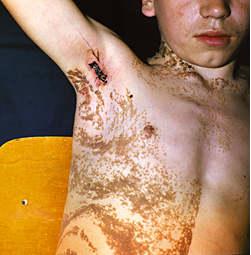
Синонимы: бородавчатый, гиперкератотический, линеарный, ихтиозиформный, твердый, односторонний невус. Термин "папилломатозный порок развития" принадлежит А. К. Апатенко (1973). Это заболевание относится к порокам развития эпидермиса и причислено в гистологической классификации ВОЗ к гамартомам, чем подчеркивается его дисэмбриогенетическое происхождение. Ранее папилломатозный порок развития рассматривался как предраковое заболевание, относящееся к опухолям поверхностного эпидермиса.
Клиническая картина характеризуется возникновением, обычно уже в первые годы жизни, бородавчатых папилломатозных разрастаний, обнаруживаемых практически на любом участке кожного покрова. В некоторых случаях появлению элементов бородавчатого невуса могут предшествовать различные агрессивные экзогенные воздействия на кожу: механическая травма, ожоги различной природы и пр. В таких ситуациях возможно первичное выявление болезни и в значительно более старшем возрасте. Половых различий в частоте встречаемости папилломатозного порока развития не наблюдается. Имеются немногочисленные свидетельства генетической и наследственной предрасположенности к возникновению этой патологии.
Различают две клинические формы папилломатозного порока развития: ограниченную и диссеминированную. В первом варианте очаги невуса единичны (от одного до нескольких) и представляют собой ограниченные образования темного цвета (от светло-серого до буро-коричневого), умеренно возвышающиеся над поверхностью кожи, располагающиеся на широком основании и имеющие на поверхности сосочковидные выросты, покрытые, роговыми наслоениями. При втором варианте число очагов в виде бляшек, разбросанных по всему телу, может достигать нескольких десятков. Отмечается определенная закономерность в топографии поражения: элементы обычно располагаются на туловище сегментарно, в зонах Захарьина-Геда либо по ходу нервных стволов и крупных сосудов (линеарный невус). Элементы не имеют склонности к инволюции и самопроизвольному разрешению, могут существовать годами без видимых изменений или увеличиваться в размере, чаще за счет увеличения толщины корок, реже за счет увеличения площади новообразования.
Папилломатозный порок развития нередко сочетается с другими пороками развития - пигментным невусом, невусом сальных желез, кистами, заболеваниями ЦНС, эпилепсией, изменениями строения скелета и пр., что подразумевает под собой в плане диагностики привлечение специалистов соответствующего профиля (онколога, рентгенолога, невропатолога).
Довольно распространенным является порок развития эпидермиса, который клинически имеет вид сгруппированных папилломатозных элементов, зачастую формирующих скопления линейной конфигурации, за что их часто именуют линейными невусами. Количество папилломатозных элементов, их размер, степень пигментации существенно варьируют от мелкоочаговых скоплений до субтотальных поражений кожного покрова .
При достаточно выраженной пигментации папилломатозные пороки развития клинически неотличимы от истинного папилломатозного невуса, т.е. содержащего невусные клетки. Чисто внешнее сходство порой является причиной чрезмерной осторожности при решении вопроса о выборе метода лечения. В ряде случаев на поверхности папилломатозных элементов имеет место усиленный гиперкератоз, что дает повод некоторым авторам рассматривать этот тип папилломатозного порока развития в качестве варианта ихтиозиформных поражений.
Невус сальных желез Ядассона
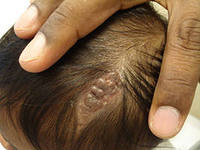
Син.: невус сальных желез — частый органоидный эпидермальный невус, обусловленный пролиферацией и мальформациией в первую очередь сальных желез, а также других компонентов кожи (эпителиальных клеток, волосяных фолликулов, соединительной ткани, апокринных желез). Описан А.Н. Mehregan и Н. Pinkus в 1965 г. Является результатом дифференцировки плюрипотентных клеток в сторону зрелых сальных и апокринных структур; именно плюрипотентной природой этих клеток объясняют развитие на его фоне других опухолей придатков кожи. Невус сальных желез в 2/3 случаев носит врожденный характер, может возникать в грудном и лишь иногда в более позднем детском возрасте. Он развивается, как правило, спорадически, хотя описаны отдельные семейные случаи. Распределение по полу одинаковое. Обычно невус сальных желез располагается на волосистой части головы (на границе с шеей, в области виска, лба) или центральной части лица, редко в других местах.
Клинически невус сальных желез Ядассона проявляется бессимптомной, солитарной, плоской, мягкой, эластичной, блестящей бляшкой овальной или линейной формы диаметром от 0.5 до 9 см или мелкими полушаровидными узелками розового, желтого, оранжевого или песочного цвета с желобоватой или несколько папилломатозной поверхностью, лишенной волос. Иногда может иметь кератотическую поверхность.
Трехстадийное течение обусловлено возрастной дифференцировкой сальных желез: 1) у младенцев и детей раннего возраста элементы невуса лишены волос, гладкие или с нежной сосочковой поверхностью; 2) в пубертатный период очаги поражения приобретают сходство с тутовой ягодой, т.к. их поверхность покрывается тесно прилегающими друг к другу полушаровидными, нередко бородавчатыми папулами от желтого до темно-коричневого цвета; 3) в зрелом возрасте примерно в 20% случаев в толще невуса развиваются доброкачественные или злокачественные опухоли, в том числе сосочковая цистаденома, гидраденома, апокринная цистаденома, кератоакантома, инфундибулома, базалиома, плоскоклеточный рак, рак апокринных желез. В частности, базалиома на фоне невуса сальных желез развивается в 5-20% случаев. При этом она возникает значительно чаще, чем доброкачественные опухоли кожи. Нередко отмечается ассоциация доброкачественных и злокачественных новообразований кожи у одного больного. Важно учитывать, что, несмотря на иногда присущую агрессивность гистологических признаков, большинство раков кожи, развившихся на фоне невуса сальных желез, отличается низкой степенью злокачественности и метастазирует лишь в исключительных случаях . Возможность развития злокачественных новообразований на фоне невуса сальных желез в раннем возрасте послужила W. Lever основанием для гипотезы о том, что развитие на фоне подобного невуса базалиомы является не злокачественной трансформацией, а результатом снижения дифференцировки первичных эпителиальных зародышевых клеток и последующего повышения их пролиферативной активности. Справедливость такого предположения подтверждается тем, что невусассоциированные базалиомы, несмотря на развитие у больных молодого возраста, обычно отличаются небольшими размерами и отсутствием признаков агрессии.
Гистологически типичные невусы сальных желез состоят из долек зрелых сальных желез в верхнем и среднем отделах дермы, отмечается также увеличение количества других эпителиальных структур (расширенных апокринных потовых желез, абортивных волосяных фолликулов). Выделяют три стадии гистологического развития невуса. 1. Ранняя стадия — проявляется гипоплазией сальных желез и волосяных фолликулов. 2. Зрелая стадия — характеризуется акантозом, папилломатозом эпидермиса, обилием гиперплазированных сальных желез, недоразвитием волосяных фолликулов, хорошим развитием апокринных желез. 3. Опухолевая стадия. Изредка встречающийся распространенный сальный невус может носить системный характер с поражением ЦНС, глаз, костной, сосудистой, мочеполовой и других систем. Очень редкий синдром сальногоневуса Ядассона включает триаду — линейный сальный невус, эпилепсю и умственную отсталость.
Дифференциальный диагноз невуса невуса сальных желез Ядассона проводят с аплазией кожи, отличающейся более гладкой папирусовидной поверхностью; сирингоцистаденоматозным сосочковым невусом, имеющим более розовую, чем желтую, узловатую, а не бархатистую поверхность; ранней ювенильной ксан-тогранулемой (быстро развивающейся куполообразной папулой или узлом) и солитарной мастоцитомой, имеющими характерное гистологическое строение; гипертрофией мозговой ткани или энцефалопатией, при которых также может определяться подкожный узел, но содержимое связано с головным мозгом. Лечение невуса сальных желез Ядассона. В связи с угрозой злокачественной трансформации принято профилактическое хирургическое удаление невуса не позже наступления раннего подросткового возраста. Электрокаутеризация и криодеструкция могут привести к рецидиву.
Пороки развития волосяных фолликулов могут иметь различные клинические проявления. Они отражают ту стадию формирования пило-себацейного комплекса, на которой процесс нормального развития был прерван по какой-либо причине. Поэтому спектр этих нарушений может включать как расположенные глубоко в дерме очаговые клеточные скопления, похожие на зародышевые почки волосяного фолликула, так и полностью сформированные терминальные волосы на ограниченном гиперпигментированном участке безволосой поверхности (невус Бекера).
Невус Беккера
(син.: меланоз Беккера, пигментный волосяной эпидермальный невус, синдром Беккера—Рейтера) — относительно частый эпидермальный невус.
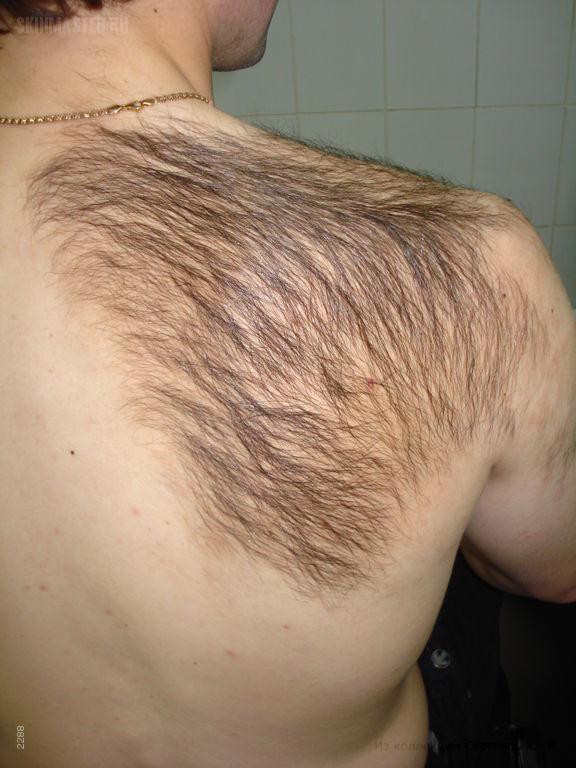
Отличается поздним возникновением — обычно в возрасте 10-15 лет, преобладанием у мужчин (в 5 раз чаще, чем у женщин), что наряду с развитием на его поверхности гипертрихоза подтверждает гипотезу о важной роли анд-рогенов в его этиологии. Становится заметным после инсоляции. Описаны семейные случаи. Вначале появляется пигментное пятно неправильной формы невуса Беккера, обычно в области плеча, под молочными железами, на спине, предплечье, голени, реже — в области таза, очень редко — на лице и шее. Как правило, пятно одностороннее, располагается вдоль линий Лангера. Пятно постепенно увеличивается, образующиеся по краям островки пигментации сливаются с основным очагом, за пределами которого появляются новые пятна что придает ему географические очертания. Элемент достигает величины 20 см или более, приобретая неправильную форму с неровными, зубчатыми границами, неравномерную окраску желто-коричневого или коричневого цвета. Вскоре после наступления половой зрелости пятно покрывается толстыми, темными волосами, но границы пигментации и гипертрихоза точно не совпадают. На некоторых участках пигментированной кожи гипертрихоз не развивается, в то же время грубые волосы покрывают внешне неизмененную кожу. Кожа становится сморщенной, утолщенной к центру элемента. Бородавчатая поверхность едва ощутима, но хорошо определяется при боковом освещении. Иногда могут быть элементы, напоминающие угри (комедоны. папулы, пустулы, кистозные узлы) . При гистологическом исследовании невуса Беккера выявляются акантоз, гиперкератоз, изредка — роговые кисты, дерма утолщена и содержит множество тяжей гладкомышечных волокон, связанных с волосяными фолликулами или кровеносными сосудами; невусные клетки отсутствуют, количество меланоцитов не увеличено, содержание меланина в кератиноцитах базального слоя повышено. При электронной микроскопии отмечено увеличение числа меланосом в клетках базального слоя эпидермиса. Меланоцитарная активность повышена, но количество меланосомальных комплексов в кератиноцитах соответствует степени повышения синтеза меланина. Гигантские меланосомы невуса Беккера обнаруживаются в меланоцитах и кератиноцитах. В очаге повышено количество андрогенных рецепторов. Течение невуса Беккера доброкачественное, без изменений на протяжении всей жизни, иногда наблюдается уменьшение интенсивности пигментации. Обычно не отмечается ассоциации с какой-либо патологией, хотя у женщин описывают одностороннюю гипоплазию молочной железы, spina bifida, укорочение верхней конечности, недоразвитие грудной клетки. Дифференциальный диагноз невуса Беккера проводится с гигантским меланоцитарным невусом, который в отличие от невуса Беккера возвышается над уровнем кожи и существует с рождения; синдромом Мак-Кьюна—Олбрайта, при котором пятно существует с рождения, окрашено равномерно и не имеет оволосения. Гипертрихоз и гиперпигментация невуса Беккера могут представлять серьезную косметическую проблему. Лечение невуса Беккера малоэффективно: дермабразия не дает выраженного эффекта, иногда используют аргоновый лазер.
. Комедоновый невус
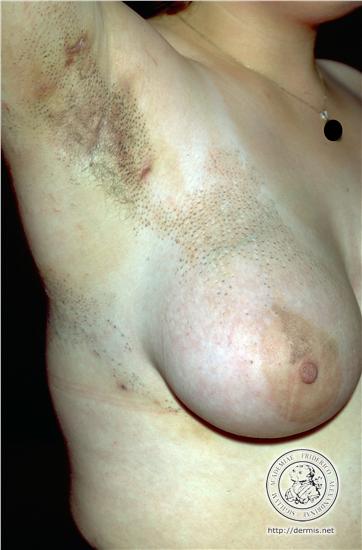
(син.: невус с комедонами, невус угревидный) — эпидермальный невус из тканей придатков кожи с множеством расширенных и заполненных кератином волосяных фолликулов. Является врожденным, иногда возникает в детском возрасте, изредка — позже. Клинически камедоновый невус представляет собой четко отграниченный очаг линейной или неправильной формы, в пределах которого имеются тесно сгруппированные, слегка возвышающиеся фолликулярные папулы. Наличие в центральной части папулы плотной роговой пробки темно-серого или черного цвета придает ей сходство с комедоном. После удаления роговой пробки остается углубление, на месте которого формируется участок атрофии. Невус может располагаться на любом участке тела. Обычно является множественным, одно- или двусторонним. При гистологическом исследовании камедоновый невус обнаруживают рудиментарные волосяные фолликулы в виде проникающих глубоко в дерму инвагинатов эпидермиса, заполненных роговыми массами, содержащих 1-2 волосяных стержня и несколько небольших атрофичных долек сальных желез: вокруг инвагинатов образуется небольшой воспалительный инфильтрат. Дифференциальный диагноз камедонового невуса проводят с бородавчатым эпидермальным невусом. проявляющимся резко выраженным гиперкератозом и бородавчатыми изменениями, отсутствующими при угревидном невусе; порокератозом Мибелли, который чаще возникает у мужчин в области конечностей, отличается крупным (до 2 см) диаметром очагов, наличием рогового валика вокруг западающего центра, отсутствием роговых пробок, имеющихся при угревидном невусе. Иногда на фоне комедонового невуса развиваются доброкачественные эккринные опухоли, трихолеммальные кисты, базалиома. Невус обычно протекает бессимптомно, но при крупных и широко распространенных очагах может потребоваться вмешательство с целью косметической коррекции. Эффективных методов лечения камедонового невуса не существует, небольшие невусы удаляют хирургически или путем электрокоагуляции, крупные — аппликациями 0,1% раствора ретиноевой кислоты.
Сирингома
Порок развития потовых желез . Клиническая картина характеризуется мелкими папулами телесного цвета, иногда с полупрозрачной передней стенкой. Характерна периорбитальная локализация, хотя последняя не является исключительной. Первые элементы обычно возникают в пубертатном периоде, но известны случаи начала процесса на фоне климакса.
Гистологическая картина характеризуется наличием множественных мелких кистозных образований в поверхностных и средних отделах дермы, стенки которых выстланы двумя рядами эпителиальных клеток, при этом те, которые обращены в просвет кисты, напоминают клетки выводных протоков эккринных потовых желез. Помимо кист, имеются тяжи из мелких темноокрашенных клеток, часть из которых связана с кистами, образуя «хвостики», что является характерным гистологическим признаком сирингомы.
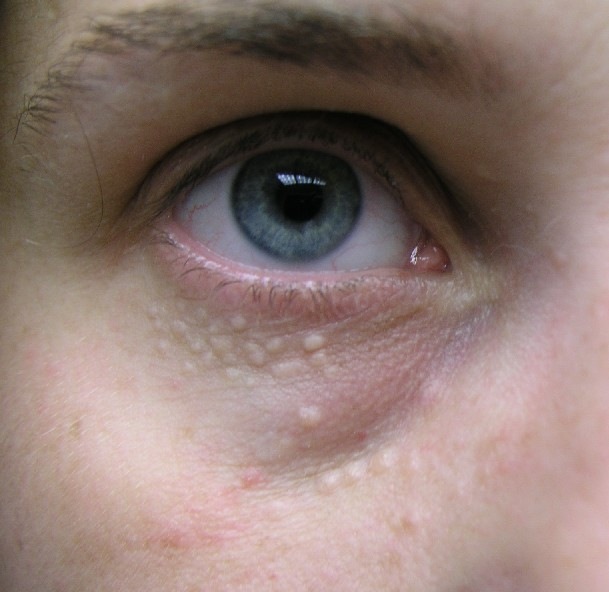
Дифференциальная диагностика
Сирингому следует дифференцировать от других опухолей потовых желез: сосочковой сирингоцистаденомы, эккринной спираденомы, а также от аденоидно-кистозной эпителиомы Брука, множественных ксантом, аденомы сальных желез Прингла, трихоэпителиомы, лейомиомы, синдрома Гольтца-Горлина.
Сосочковая сирингоцистаденома отличается от сирингомы в клиническом отношении тем, что в большинстве случаев является солитарным, а не множественным новообразованием и локализуется преимущественно на волосистой части головы, в паховых и подмышечных складках, а не на лице, шее и туловище, как сирингома. Элементы сирингоцистаденомы в отличие от сирингомы весьма вариабельны и могут быть подобны узелкам, бляшкам с папиллярными выростами, кистозным элементам. Гистологически сирингоцистаденома отличается от сирингомы отсутствием множественных кист в дерме, папилломатозом эпидермиса и густым плазмоцитарным инфильтратом.
Эккринная спираденома в отличие от сирингомы чаще возникает у мужчин, имеет вид солидарной опухоли (реже представлена множественными узлами, глубоко залегающими в дерме) плотной консистенции, болезненна при пальпации. Кожа над узлами не изменена или имеет цианотическую окраску. Гистологически эккринная спираденома отличается от сирингомы отсутствием множественных кистозных образований в дерме, формированием элементов дольчатого строения, инкапсулированных, не имеющих связи с истонченным эпидермисом.
Аденоидно-кистозная эпителиома Брука, так же как и сирингома, чаще возникает у женщин молодого возраста, характеризуется множественными очагами поражения, но в отличие от сирингомы может иметь семейный характер. Элементы при аденоидно-кистозной эпителиоме могут появляться на конечностях, группироваться. Они имеют гладкую поверхность, в ряде случаев с выраженными телеангиэктазиями, могут сочетаться с сирингомой, цилиндромой и другими новообразованиями кожи. Гистологически аденоидно-кистозная эпителиома отличается от сирингомы наличием структур, напоминающих волосяные фолликулы.
Множественная узелковая ксантома отличается от сирингомы прежде всего желтоватой или желтовато-оранжевой окраской элементов, для которых характерна излюбленная локализация в периорбитальной области, на шее и туловище, как это свойственно сирингоме. Кроме того, множественная узелковая ксантома в отличие от сирингомы не возникает преимущественно у женщин. Гистологически она характеризуется наличием ксантомных клеток и отличается от сирингомы отсутствием кистозных образований в дерме.
Клинические исследования
Проведенные клинические исследования доказывают высокую эффективность, безопасность и переносимость продукции ТМ ««Ла-Кри». Средства подходят для ежедневного ухода за взрослой и детской кожей с легкой и средней формой атопического дерматита и в период ремиссии. В результате терапии отмечено снижение активности воспалительного процесса, уменьшение сухости, зуда и шелушения.
Средства рекомендованы союзом педиатров России.
Источники:
- Молочкова Юлия Владимировна, Дерматология. Краткий справочник, ГЭОТАР-Медиа, 2017.
- Горланов И. А., Леина Л. М.,Милявская И. Р., Заславский Д. В., Болезни кожи новорожденных и грудных детей. Руководство для врачей, СПб.: Фолиант. 2016.
- Суколин Геннадий Иванович, Иллюстрированная клиническая дерматология. Краткий алфавитный справочник, изд-во Люкс Принт, 2010.








Читайте также

